

This podcast, part two of a two-part series, discusses practical tips on how to incorporate cybersecurity into your own practices and devices.

This MAPS podcast details what medical device cybersecurity is and why you should care.

This MAPS podcast explores ways medical device companies can capitalize on the shift from inpatient surgical settings to hospital outpatient departments.

Download the full article here
By
Ajit Patwardhan, MD1, MBA; Robin Winter-Sperry, MD2; Dannielle Heuer3; Stephen Valerio4; Urvashi Vashee5; Drilon Saliu, PharmD, MBA6; Greg Christopherson, PhD7; Irma Saliu, PharmD8; Kyle Kennedy9; Michelle Powell, PharmD10; Chris Brock, PharmD11 ; John B Pracyk, MD, PhD, MBA12; Joe Medicis, PharmD13
1 Physician – Surgeon, Medical Affairs Executive – Baxter International, Integra LifeSciences, NLT Spine, Paradigm Spine
2 President, Scientific Resilience; MAPS 2020 Co-Chair
3 Director WD Communications
4 Sr Director, Oncology Medical Training AstraZeneca
5 Global Scientific Training Merck & Co
6 Head, Global Medical Affairs, Clinical Development and HEOR, Connected Care Philips
7 VP, Medical Affairs Medline
8 National Director, Psychiatry MSLs, US Field Medical Allergan
9 VP, Customer Strategy – The Medical Affairs Company
10 Director, Filed Medical Excellence Astellas Medical Affairs Americas USA
11 Field Based Medical Affairs, Head, Respiratory GlaxoSmithKline
12 Integrated Leader, Medical Affairs, Pre-Clinical, & Clinical Research DePuy Synthes Spine, (J&J)
13 Director US Medical Affairs Callidiatas Therapeutics

The evolving role of Medical Sciences Liaisons (MSLs) was extensively discussed in multiple presentations and workshops at the March 2020 Medical Affairs Professional Society (MAPS) Global Annual Meeting in Miami, Florida. Executive leaders with domain expertise in building and managing Field Medical teams shared their experience and views on the value proposition of these roles and how a changing healthcare and industry landscape is influencing differentiation of and new opportunities for this role. The leaders also recognized the increased need for specialized training and how adult learning principles along with integration of new creative digital tools could help enhance engagement, performance and career growth. This article formalizes these learnings, providing best practices for MSLs in the context of the ongoing pandemic and beyond.
MSLs represent a core function of Medical Affairs, acting as the medical face of the organization to provide deep sub-specialty knowledge to healthcare providers and other external stakeholders. MSLs’ primary responsibility focuses on the three pillars of Medical Affairs, i.e. Scientific Exchange, Evidence Generation, and Evidence Dissemination. As such, a primary function of the MSL role is to build and execute an engagement plan in an expanding thought-leadership network. Currently, both the development of these plans and the actions that allow MSLs to deliver on their promise are undergoing sea change.
Over the last decade, healthcare delivery has undergone sweeping reforms. Emerging trends now focus on substantially better cost, quality, and outcomes as the new parameters to demonstrate significant healthcare value. Patients are now at the center of making their healthcare decisions and are demanding data transparency, easy/convenient access, and personalized products and services. Increased need for data transparency and availability has given rise to disruptive technologies and technological advances in the form of advanced data analytics, artificial intelligence, machine learning, digitization, new social media platforms, etc. Alongside these societal and cultural changes, stricter regulations and adjustments to healthcare policy (e.g. changing insurance landscape, use of Real World Evidence, new European Union (EU) regulations, increased scrutiny on drug pricing and rebates, etc.) are driving new ways of thought and action in the healthcare landscape. Meanwhile, the promise of new technologies driving basic and translational research have led Pharmaceuticals and MedTech industries to renew their focus on R&D and scientific advances, leading to personalized medicine breakthroughs in drug-device combinations, small molecule therapeutics, biologics, immunotherapy and diagnostic biomarkers, regenerative medicine and many more areas of advancement in the life sciences industry.
With these dynamic changes taking place in the healthcare landscape, the traditional role of an MSL as a bridge between internal company stakeholders and the outside medical community now needs to expand to encompass new competitive skillsets focusing broadly on Products, People, Personal & Platforms (“4Ps”). The following description of these 4Ps is not meant to be comprehensive, but rather is an attempt to start a new conversation about these core areas to better equip MSLs with a forward-looking understanding of the skill sets needed to succeed in the current and future disrupted healthcare landscape.
Due to increased need for personalized medicine (as described in part above), complex treatment options require deep therapeutic area expertise, which for MSLs includes the following:
The model of proscriptive medicine in which treatment decisions were made almost solely by healthcare providers has given way to a new model in which patients are collaborators or even drivers of treatment choices, often taking into account quality of life alongside quantity of life. This shift results in new stakeholders, including but certainly not limited to the following:
Along with new Products and new People are new Platforms that offer significant opportunity for MSLs to reach stakeholders with new creative digital tools for scientific engagement and education, including the following:
The paradigm shift in the industry, requires MSLs to adapt their practices through specialized training including continuous learning for role proficiency, along with enhanced performance and career growth. The following considerations can guide organizations and individuals in their development of training programs and curricula. MSL training should be:
Furthermore, trainings designed to help MSLs build the skillsets needed to keep pace with changing market, social, technological and regulatory conditions should include assessment and certification requirements, often including the following:
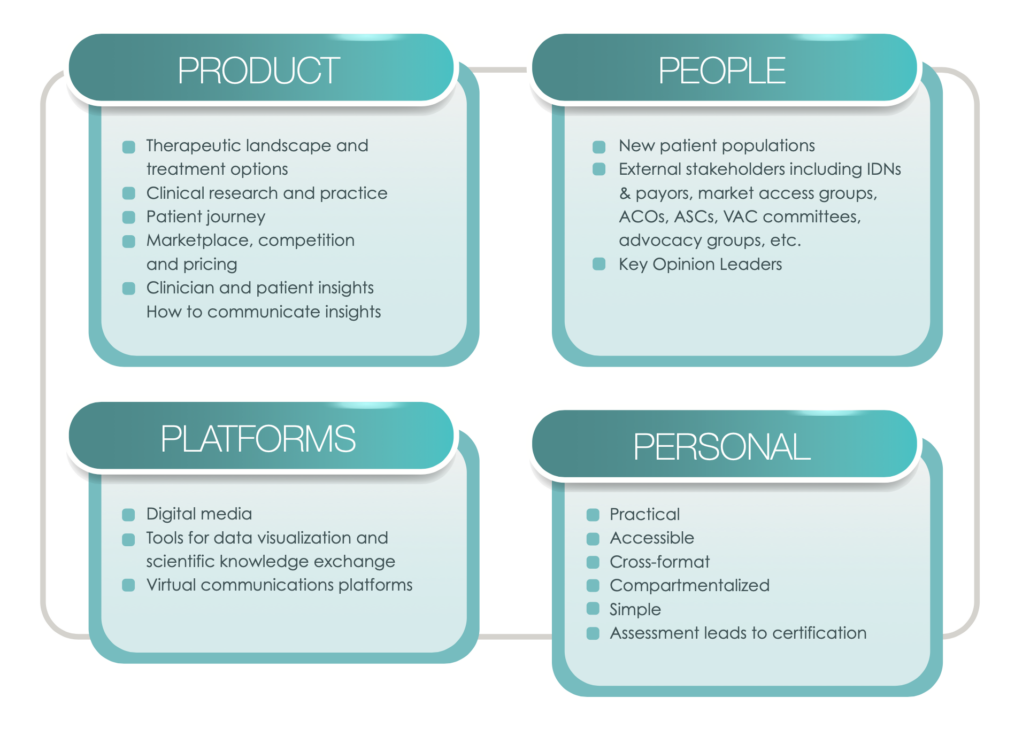
The Four 4 Ps: Considerations for MSL Training Design
Innovations in science, data and digitization have transformed the healthcare landscape, leading to the emergence of new decision-makers and stakeholders. Alongside these innovations are shifts in culture and society that see patients playing a more active role in their treatments. These shifts are not hypothetical – they are here. The evolving healthcare environment presents both opportunity and challenge for MSLs to further differentiate their competencies, for example through specialized understanding of the therapeutic landscape, healthcare networks, and the requirements and desires of a diverse population of stakeholders. Through proactive engagement with new learnings, MSLs have the opportunity to broaden their vision, role and influence. These new skills will allow tomorrow’s MSLs to elevate performance across medical engagements to optimize experiences and outcomes for physicians and patients.
This publication represents the consensus opinions of the authors and various members of MAPS, but does not represent formal endorsement of conclusions by their organizations

Download the Full Article Here
By João D. Dias1, Cesar Sanz Rodriguez2, Raphael T.B. Tan3
1 Medical Affairs & Clinical Development, Haemonetics S.A., Switzerland, Medical Affairs Professional Society (MAPS) MedTech Focus Area Working Group (FAWG) EMEA Co-Lead
2 Medical Affairs, MSD International GmbH, Switzerland, MAPS MedTech FAWG EMEA Co-Lead
3 International Medical Executive Consultants (IMEC), The Netherlands, MAPS MedTech FAWG member

This article provides an overview of the new MedTech European regulatory environment and opportunities for the Medical Affairs function to evolve and bring value to the respective organizations. The European Regulations ask for an increased effort from manufacturers to generate and communicate clinical evidence on the safety and performance of their Medical devices, In vitro-diagnostics and Drug Device Combinations. In conjunction with increased quality standards they make a compelling case for Medical & Scientific Governance with a prominent role for Medical Affairs in many pre- and post-market processes. A transformation of Medical Affairs into a strategic business function makes the medical device industry an exciting place to be for Medical Affairs professionals.
For over two decades, Medical devices and In-vitro Diagnostics have been regulated in Europe by Directives for MEDICAL DEVICES (MDD) and IN VITRO DIAGNOSTICS (IVDD)1,2,3 that were published in 1993 and 1998 respectively. A separate Directive for Active Implantable Medical Devices (AIMD) was published in 1990 with a last revision in 2009. According to these Directives, devices are approved for the European Single Market only after having obtained CE Mark for which manufacturers need to demonstrate conformity to essential requirements relating to the device’s performance and safety for patients and users. After public consultation by the European Commission in 2008 it became clear that an update of the Directives was needed, one reason being the simple fact that new technologies such as companion diagnostic devices were not yet covered. The need for revision gained traction after incidents with breast implants, transvaginal meshes around 2009 and metal-on-metal hip prostheses a couple of years later. Eventually, the revision process that started in 2012 resulted in the Medical Device Regulation (MDR) in which MDD and AIMD were combined and the In Vitro Diagnostics Regulation (IVDR)4,5. The new Regulations were published in 2017, with May 25, 2017 as the official date of entering into force. A transition period to full implementation of the MDR and IVDR was allowed for three and five years respectively, which means the MDR applies from May 26, 2020 and IVDR from May 26, 2022.
So far so good… and then the COVID-19 crisis struck Europe, right at the moment when the medical device industry and notified bodies are transitioning to the new Regulations. Therefore, in order to “take the pressure off national authorities, notified bodies, manufacturers and other actors so they can focus fully on urgent priorities related to the coronavirus,” the European Commission has decided to move back the date on which the new MDR would fully apply by one year, to 26 May 2021. MedTech Europe, the European trade organization of medical device manufacturers, has advocated for a similar delay for the IVDR.
Since the early days of the global COVID-19 crisis the medical industry has made a so far unseen effort in finding (bio-)pharmaceutical solutions, developing vaccines and reliable test kits. Simultaneously, in an attempt to address the relative shortage of masks and intensive care equipment such as ventilators, traditional medical device manufacturers ramped up production. Although regulators accommodate the surge of new devices by fast tracks and exemption rules, new devices are subject to meticulous assessments of performance and safety. And rightly so, since national policies to curb transmission of the virus rely on the quality of diagnostics and personal protection equipment. Especially important is the scrutiny in assessing new medical devices which are intended to be used in the management of the most vulnerable and severely ill COVID-19 patients who end up in hospitals and ICUs.
In general the new Regulations devices, improve traceability and transparency and define stricter requirements to clinical evidence and post-market surveillance. The MDR now also regulates devices for cosmetic purposes such as colored contact lenses and cosmetic implant devices. The introduction of Unique Device Identification should improve traceability, and transparency is created by the European Databank of Medical Devices (EUDAMED) where all mandatory regulatory documentation on each device is kept and updated. Manufacturers must re-certify devices in accordance with the new regulations and update their technical documentation accordingly with special attention to higher clinical requirements for class III and implantable devices. A shift of focus from a mere pre-market perspective towards a life-cycle approach also includes stricter requirements regarding post-market surveillance, post-market clinical follow-up and vigilance.
This brings us to the Clinical Evaluation, which lies at the basis of CE mark approval and the life-cycle approach. It is defined as “a systematic and planned process to continuously generate, collect, analyze and assess the clinical data pertaining to a device in order to verify the safety performance, clinical benefits of the device when used as intended by the manufacturer.” The Clinical Evaluation process is described in a MEDDEV (MEDical DEVices) guidance document. MEDDEVs promote a common approach to be followed by manufacturers and notified bodies that are involved in conformity assessment procedures. Although these MEDDEVs are not legally binding, it is expected that their guidance be followed, ensuring the uniform application of the various elements of the directives/regulations. MEDDEV 2.7/1 rev. 46 is the guidance document with regards to Clinical Evaluation and is prescriptive on the process, the required qualification of the evaluators and the contents of a Clinical Evaluation Report (CER).
The CER should “describe the intended clinical benefit and provide evidence of safety and performance” where performance is defined as “the ability of a device to achieve its intended purpose as stated by the manufacturer.” This report describes the risk profile of the device based on the technical documentation and provides an appraisal of all available clinical data related to safety and performance. Any evidence gaps and residual risk need to be addressed in Post-Market Clinical Follow Up (PMCF) studies to demonstrate long-term performance safety. The results from the intensified surveillance are laid down in the Periodic Safety Update Report (PSUR, mandatory for Class IIa/b and III) and Summary of Safety and Clinical Performance (SSCP, for implantables and Class III). These documents must be uploaded in EUDAMED, which allows public access to the SSCP.
Although European market approval of new devices can still be obtained by referring to clinical data of predicate equivalent devices, the MDR explicitly lists criteria by which equivalence can be claimed from a technical, biological and clinical perspective. The equivalence under the MDD was less well defined. Under the MDR, the predicate device must have a similar design, use the same materials and come in contact with the same tissues and body fluids, and be used for the same clinical indications. If a device does not meet these criteria, manufacturers need to generate their own clinical evidence with appropriate clinical investigations.
CE Marking requires Notified Body involvement for most medical device classes and is related to implementation of a Quality Management System, which must include plans pertaining to Clinical Evaluation, Post-Market Surveillance (PMS) and Post Market Clinical Follow-up (PMCF). For non-sterile Class I devices, manufacturers can conduct the conformity assessment themselves and basically self-certify. For Class I devices that are sterile, measuring or reusable surgical instruments as well as for all higher device classes, oversight of a Notified Body is required. In the end, it is the Notified Body that issues a CE Marking Certificate and ISO 13485 Certification. Only with these certificates in place the manufacturer can prepare a Declaration of Conformity and put the CE Mark sign on their products and labelling.
In Vitro diagnostics (IVDs) are medical devices with which tests are performed using human specimens such as urine or blood. Familiar examples of such devices are pregnancy tests and tests for determining the level of glucose or cholesterol in the blood. The current directives distinguish between medium risk and high-risk lists of IVDs. Those IVDs not captured in these lists are automatically classified as low risk. Obviously more stringent market authorization procedures with Notified Body oversight apply to high risk IVDs. Whereas in the past only a minority of IVDs required involvement of a Notified Body, under the new classification an estimated 90% will now require it. The IVDD had some gaps and this binary system was thought to be no longer sufficient. Hence, like for the MDR, a risk-based approach classification was introduced based on the severity of the disorder tested for and possible consequences of an incorrect test result. Instead of two lists, the new IVDR now distinguishes four categories, Class A (lowest risk), Class B, Class C, and Class D (highest risk) and dictates that Class B and above IVDs will require oversight from a Notified Body as part of their conformity assessment.
Aside from the risk-based classification, it will not surprise you that the new IVDR has more features in common with the MDR. As is the case for medical devices, the IVDR requires clinical evidence and post-market performance follow-up. This will require a Performance Evaluation plan and report for all IVD Classes, which will describe how to demonstrate scientific validity, analytic performance, and clinical performance.
Some medicines are used in combination with a medical device, usually to enable the delivery of the medicine. In the European Medicines Agency (EMA) view, if the principle intended action of the combination product is achieved by the medicine, the entire product is regulated as a medicinal product under Directive 2001/83/EC7 or Regulation (EC) No 726/20048. However, MDR’s Article 117 brought some relevant additions. First, two categories were defined: (a) Integral, where the medicinal product and the device form a single integrated product (e.g. pre-filled syringes and pens) and (b) Co-packaged, where the medicinal product and the device are separate items contained in the same pack (e.g. reusable pen for insulin cartridges). Next, Article 117 also incorporated some relevant amendments to Directive 2001/83/EC to ensure combination products comply with the medical device legislation. Per MDR’s Article 117, the marketing authorization application should include a CE certificate for the device or an opinion from a Notified Body on the conformity of the device (except for non-sterile, non-measuring and non-reusable surgical Class I devices).
Probably as a reaction to the rapid growth of combination products in recent years and the need to bring further clarity in this area, in June 2019 EMA released for public consultation a draft Guideline on Quality Requirements for Regulatory Submissions for Drug-Device Combinations9. The aim of this Guideline is to clarify expectations laid down in Directive 2001/83/EC and address the new obligations in the MDR. EMA makes it clear that the Notified Body assessment and marketing authorization review would not result in duplicate assessments. The former will review the device alone, while the latter will ensure the safety and efficacy of the drug are not compromised by the inclusion of the device part. The consultation period ended in August 2019 and EMA is now due to finalize the Guideline in the second quarter of 2020.
It is also worthwhile making a reference to medical devices which may contain an ancillary medicinal substance to support the proper functioning of the device (e.g. drug-eluting stents). These products should comply with the medical device legislation. Yet, the manufacturer should also seek a scientific opinion from EMA on the quality and safety of the ancillary substance if it is derived from human blood or human plasma, or if it is within the scope of the centralized procedure for the authorization of medicines. For other substances, the Notified Body can seek the opinion from EMA or a national competent authority. Of note, EMA has recently issued a Consultation Procedure for Ancillary Medicinal Substances in Medical Devices.
Companion diagnostics are seen by EMA as in vitro diagnostic tests that support the safe and effective use of a specific medicinal product, by identifying patients that are suitable or unsuitable for treatment. Applicable regulations were discussed above. Yet, before the Notified Body can issue CE Marking, it must seek a scientific opinion on the suitability of the companion diagnostic to the medicinal product concerned from EMA or a national competent authority, as appropriate. Similarly, a scientific opinion would also be needed for some other medical devices made of substances that are absorbed by the human body to achieve their intended purpose. These devices are normally introduced into the human body via an orifice or applied to the skin. Last, we should not forget the so called “borderline products”. These are complex healthcare products for which there is uncertainty over which regulatory framework applies. Common borderlines are between medicinal products, medical devices, cosmetics, biocidal products, herbal medicines and food supplements. The European Commission publishes the ‘Manual on borderline and classification in the Community regulatory framework for medical devices’ which provides examples and recommendations for determination of classifications. National competent authorities classify borderline products either as medicinal products or, for example, as medical devices on a case-by-case basis based on the product’s composition and constituents, its mode of action and its intended purpose. This determines the applicable regulatory framework.
The new European regulatory environment (MDR, IVDR and EMA guidance on Drug Device Combinations) in conjunction with expanded quality standards for MedTech products and a rapidly changing reimbursement landscape are intensifying the need for a Medical Affairs role in the product lifecycle management. Furthermore, the increased trade organization’s guidelines as well as a more complex competitive environment in which the direct comparator might not be another MedTech product but instead a Drug or a Drug Device Combinations, all work favorably for Medical Affairs to step up its game and demonstrate its value in many regards.
The traditional competencies of Medical Affairs are still required to produce the new mandatory regulatory deliverables (Clinical/Performance Evaluation Report, Periodic Safety Update Report, Summary of Safety and Clinical Performance). However, more than ever Medical Affairs involvement needs to be formalized for various other key processes ranging from product development to device application. In order to give real meaning to patient and customer centricity, the inclusion of Medical Affairs contribution is indispensable with regard to, for instance, risk analyses, claims development and identifying user training needs.
In addition, medical and clinical research functions must lead in the development of coherent and affordable clinical research programs that serve regulatory compliance, reimbursement and commercial adoption purposes. The increased effort the companies must make to generate clinical evidence will undoubtedly put strain on human and financial resources. The careful planning and design of studies should avoid waste in time and money while providing the evidence the business needs in a timely fashion. Therefore, as non-inferiority assessments are giving space to superiority assessments, an increased is expected in reliance for regulatory purposes on less conventional evidence generation strategies such as registries, collaborative research and investigator lead studies.
Furthermore, ensuring the safe application of current devices, medical communication and interactions with healthcare stakeholders and health authorities, collecting and weighing medical intelligence on future directions in healthcare all require medical scientific oversight. The medical scientific oversight will be fundamental in the advancement of the company’s innovation agenda as the bar is higher than ever, as are the associated entry and maintenance costs.
The new environment makes a compelling case for installing a structure for Medical and Scientific Governance and a proactive strategic role for Medical Affairs. This in turn opens the discussion on how to develop and organize competencies around organizational capabilities that relate to the development of innovative and relevant devices, the substantiation of medical-clinical and health economic claims and ultimately oversight to ensure safe and appropriate use. Thus, we argue that now is the time to engage in captivating thought exercises about whether a company’s fabric with a prominent role of Medical Affairs adds to the organizational capability and resilience to handle current and future challenges.
This publication represents the consensus opinions of the authors and various members of MAPS, but does not represent formal endorsement of conclusions by their organizations
1. EU Council Directive 93/42/EEC of 14 June 1993 concerning medical devices. EUR-Lex; an official website of the European Union. Published July 12, 1993. Accessed August 27 2020.
2. EU Council Directive 98/79/EC concerning in-vitro diagnostic medical devices. EUR-Lex. Published December 7, 1998. Accessed August 27, 2020.
3. EU Council Directive 90/385/EEC of 20 June 1990 on the approximation of the laws of the Member States relating to active implantable medical devices, EUR-Lex, Published July 20, 1990. Accessed August 27, 2020
4. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. EUR-Lex. Published May 5, 2017. Accessed Aug 27, 2020.
5. Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices. EUR-Lex. Published May 5, 2017. Accessed Aug 27, 2020.
6. European Commission. MEDDEV 2.7/1 rev. 4 – Clinical Evaluation: A guide for manufacturers and notified bodies. Website of the European Commission. Released June 2016. Accessed August 27, 2020.
7. EU Council Directive 2001/83/EC of 6 November 2001 on the Community code relating to medicinal products for human use. EUR-Lex. Published Nov 28, 2001. Accessed Aug 27, 2020.
8. EU Regulation (EC) No 726/2004 of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. EUR-Lex. Published April 30, 2004. Accessed Aug 27, 2020.
9. European Medicines Agency – draft guideline on the quality requirements for drug-device combinations. EMA Website. Published June 3, 2019. Accessed Aug 27, 2020.

Click a thumbnail below to listen to the episode.

Episode 1: Host John Pracyk, MD, PhD, MBA, Worldwide Integrated Leader, Medical Affairs, Pre-Clinical & Clinical Research, DePuy Synthes – Spine | Johnson & Johnson Medical Devices Companies welcomes Philip Desjardins, JD, Vice President, Global Regulatory Affairs Spine and Medical Device Regulatory Policy, DePuy Synthes | Johnson & Johnson Medical Devices Companies, and Drilon Saliu, PharmD, MBA, Head, Medical, Clinical and HEOR, Connected Care, Philips, to share insights into the importance of compliance with the new European Device Medical Regulation (EUMDR), including key deadlines for compliance.

Episode 2: Host John Pracyk, MD, PhD, MBA, Worldwide Integrated Leader, Medical Affairs, Pre-Clinical & Clinical Research, DePuy Synthes – Spine | Johnson & Johnson Medical Devices Companies welcomes Philip Desjardins, JD, Vice President, Global Regulatory Affairs Spine and Medical Device Regulatory Policy, DePuy Synthes | Johnson & Johnson Medical Devices Companies, and Drilon Saliu, PharmD, MBA, Head, Medical, Clinical and HEOR, Connected Care, Philips, to continue their comprehensive discussion of: Covid’s impact on EUMDR; USFDA and EUMDR global regulator comparison; short and long term impacts; how Brexit might impact the UK’s regulatory oversight; the importance of investing in top talent; new capabilities that are required; the importance of leading with influence; and, evidentiary compliance planning and budget impact.
CLICK HERE to subscribe to the Elevate Podcast Channel on Apple iTunes.

Click a thumbnail below to listen to the episode.
 .
. 
 .
. 
 .
. 
CLICK HERE to subscribe to the Elevate Podcast Channel on Apple iTunes.

Medical devices and pharma: two sides of the same coin; significantly different but part of the same larger ecosystem. Dr. John Pracyk discusses the implications for Medical Affairs professionals.
So why have we called this piece Medical Devices are from Mars; Pharmaceuticals are from Venus? According to Dr. John Pracyk, it is a useful metaphor based on the classic relationship guide by Dr. John Gray: Men Are from Mars, Women are from Venus. Expanding on the analogy, he explains that the majority of Medical Affairs professionals in the medical device sphere are surgeons, whereas pharma is dominated by physicians. Beyond this, of course, there are other significant differences that stem from a close alignment between medical devices and therapeutic expertise.
 Dr. Pracyk, who is Worldwide Integrated Leader, Medical Affairs, Pre-Clinical & Clinical Research, for DePuy Synthes – Spine (part of the Johnson & Johnson stable), says: “It’s important because Medical Affairs is now aligning towards the fact that you’re most likely going to be working in an operating company in line with your sub-specialty training and board certification, along with operative proficiency and clinical practice experience, which will be at least a decade or more. After that you begin to have the skill set that industry is looking for. That’s decidedly different than Medical Affairs in pharmaceuticals, where there is a much more diverse range of clinical experience and educational backgrounds.
Dr. Pracyk, who is Worldwide Integrated Leader, Medical Affairs, Pre-Clinical & Clinical Research, for DePuy Synthes – Spine (part of the Johnson & Johnson stable), says: “It’s important because Medical Affairs is now aligning towards the fact that you’re most likely going to be working in an operating company in line with your sub-specialty training and board certification, along with operative proficiency and clinical practice experience, which will be at least a decade or more. After that you begin to have the skill set that industry is looking for. That’s decidedly different than Medical Affairs in pharmaceuticals, where there is a much more diverse range of clinical experience and educational backgrounds.
“In other words, to deliver the most value to the device industry we can only make the transition once we are board certified and bring a wealth of clinical practice experience to the table.”
Meanwhile, in pharmaceuticals, the classical model is an internal medicine residency with sub-specialty fellowship, or research training, followed by a highly variable period of time in clinical practice. Some physicians forgo residency and clinical practice entirely and make the transition to industry immediately following medical school or, alternatively, a post-doctoral research fellowship, while others have had meaningful careers in healthcare consulting.
The differences between devices and pharmaceuticals is especially significant from the perspective of the new European Medical Device Regulation that was passed on May 26, 2017 and comes into full effect on May 26, 2020, after a three-year phase-in period.
“Medical device companies need medical directors that have specific subject-matter surgical expertise that reads directly to the product portfolio. Why? Because the MDR credentials section has become more stringent, as have the state-of-the-art requirements – these require medical affairs surgeons to have deep clinical insights into a specific therapeutic area, as they are medically responsible as signatories for the Clinical Evaluation Report, (CER) which is the formal application for the CE mark in the European Union. For example, as a spine fellowship-trained neurological surgeon, it is quite appropriate that I lead medical and clinical affairs for our spine platform. Similarly, orthopedic surgeons lead trauma and joint reconstruction, whereas an otolaryngologist leads our ENT platform.”
Beyond this, there are further differences between devices and pharmaceuticals in terms of Medical Affairs’ scope of responsibility. “Typically, Medical Affairs in pharmaceuticals is primarily involved once the drug has launched. Clinical Affairs is responsible for all the pre-launch activities such as R&D and drug development. In devices, Medical Affairs spans that entire spectrum from initial ideation and front-end conceptualization through the stage-gate, development process, bringing that product to market and then launching it. It doesn’t stop there, as once it is in the field, maintaining it through its lifecycle and then removing from the market is also our responsibility. Medical Affairs in devices is subdivided into two major divisions, 1) Franchise, which is responsible for product development through the R&D pipeline all the way up to launch, and 2) Lifecycle, which picks up the product at launch and manages it through its entire lifespan on the market. We typically refer to it as ‘pipeline and portfolio ’: pipeline being franchise, portfolio being lifecycle.
“In pharmaceuticals, Medical Affairs is predominantly involved in the post-launch affairs, getting the product to market, medical communications, scientific engagements with KOLs, for purposes of studies, clinical trials, publications, podium presentations, and supervising all of the field medical professionals, such as medical science liaisons (MSLs).
“In devices, we are both internally and externally facing. For example, we work closely with Health Economics & Market Access (HEMA), to establish evidence of differentiated value to position products to successfully navigate value analysis committees (VACs). Similarly, we often oversee clinical investigational studies and investigator-initiated studies that support our products for purposes of safety and performance for our regulatory authorities. Internally, we work with R&D, Quality, Regulatory, Global and Regional Marketing in helping our colleagues understand what it’s like on the hospital and surgeon side of the equation.
“When I was practicing, I was the surgeon champion on the VAC for my health system. Now, I use that prior clinical and hospital administrative experience to help guide our interactions as a device manufacturer with the VACs of our customer hospitals and integrated delivery systems. Simply put, it is different ends of one vary large supply chain. Not surprisingly, I have also been involved in engaging our own contract manufacturers through speaking engagements and moderating panels at the Orthopedic Manufacturers Technology Exposition and Conference (OMTEC), as I now need to learn about how supply chains in industry work, while reciprocally informing our contract manufacturers on the global transition taking place clinically from volume to value that ultimately impacts their businesses as well.
Medical Affairs informs strategic decision-making
Medical Affairs is increasingly becoming a strategic partner for both commercial and R&D teams. “One way to look at this is through a go-to-market strategy that is refracted through the lens of three evidentiary audiences. The first is regulatory, where we must place the product ‘in country’. Next is hospitals/integrated delivery networks, where were we need to work with the VACs to get the product ‘on contract ’. Thirdly, we must secure reimbursement from the payers, whether they be private, commercial insurers, or government entities. As you can see, Medical Affairs is involved in a very broad range of medical and scientific engagement activities.
Diverse skillsets
Medical Affairs professionals in devices are fewer in number and require a very diverse skillset, whereas Medical Affairs in pharmaceuticals are far larger in number and, historically have been around much longer. “When you consider the sheer numbers of Medial Affairs in pharmaceuticals, both internally and with the MSLs in the field, it is literally an army. Conversely, on the devices side, we’re more of a ‘special forces’ model, where you just have a few people who are very specialized, but cross-functionally trained in diverse areas, which is a force multiplier.
So how hard is it to obtain the specialist knowledge and overcome the significant shortage of talent? Surgeons coming into Medical Affairs often have skillsets that are much in demand, beyond their specialist clinical expertise. For example, time spent learning how to develop a practice and building business acumen are highly prized in industry.
“Many physicians and surgeons are now going to business school to get their MBAs, while others have been involved in hospital leadership committees or have commercialized devices as an entrepreneur. These transferrable skills are valuable when you consider the range of activities that Medical Affairs delivers against: namely, understanding business operations, manufacturing, finance, accounting, communications, strategic development, and pre-clinical and clinical research – and being able to navigate the spectrum from bench top, to small animal, large animal, and ultimately first in human. Not surprisingly, surgeons who possess these essential business, research and clinical skills will meet with great success in devices.”
Leaving clinical practice
In pharma it is not uncommon for clinicians to maintain some form of clinical practice, which can take a variety of forms from an occasional clinic, to volunteering, or mission work. However, due to both legal and healthcare compliance issues, surgeons working in devices must stop operating and close their clinical practice upon entering industry. For a surgeon, who has spent years acquiring a unique set of skills this is a huge deal. Fortunately, maintenance of operative skills is accomplished through a variety of cadaver settings: wet labs, validation labs, and prototype testing.
Yet, there is one remarkable upside, according to Dr. Pracyk. “Believe it or not, we can secure visiting professor privileges anywhere in the world that permits us to scrub into surgery with key opinion leader surgeons – not to clinically perform the operation (in fact, we are specifically prohibited from touching the patient), but more importantly to observe, learn, and distill out the critical insights that help identify and address true unmet needs. The simple fact that this worldwide peer-to-peer surgeon exchange occurs from within the operative field is absolutely amazing.”
Lessons on both sides
“I think what pharmaceuticals can learn from the device side is that we are very good at these cross-functional skills.” Flipping it around the other way, what can devices learn from pharma? “I think pharmaceuticals has a much more thorough and deep understanding of ‘patient centricity’.
“In devices, the surgeon is the proxy for the patient. However, with the concept of surgeon employment and the advent of spine and brain institutes in neurological surgery or musculoskeletal institutes in orthopedic surgery, the multidisciplinary model of care is taking hold. Ultimately, the patient will be at the center of what we do and a more comprehensive approach to care redesign will certainly elevate patient centricity globally as the medical devices sector continues to learn, share and advance care more holistically like our pharmaceutical colleagues.

